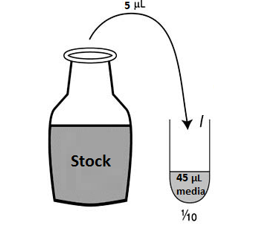
Day 20: Analysing the effect of Oxygen glucose deprivation on C17.2 cells upon the treatment of peptides TAT, Myr-p110, and TAT-p110 Aim: to assess the affect of OGD on C17.2 cells after 1.5 hours and 3 hours by assaying LDH, Hoechst and MTT. Reason for assaying LDH: LDH is lactate dehydrogenase which catalyses the conversion of pyruvate -> lactate during anaerobic respiration. Anaerobic respiration only involves the process of glycolysis which occurs only in the cytosol. Thus, if the peptides became cytotoxic to the cells, then the lactate dehydrogenase would leak out into the medium and therefore be detectable by increased absorbance and less transmission. Reason for assaying Hoechst: Hoechst specifically stains nuclei blue. If there are more nuclei present this means that less cells have died as a result of cytotoxicity. If there are less nuclei present this means that more cells have died as a result of cytotoxicity. It is required that we make a blank and then a ...